![[ N° CAS 5402-55-1 ] 2-thiophèneéthanol Image vedette](https://www.ohlsenwin.com/uploads/p1.png)
![[ N° CAS 5402-55-1 ] 2-thiophèneéthanol](https://www.ohlsenwin.com/uploads/product-2.png)
![[ N° CAS 5402-55-1 ] 2-thiophèneéthanol](https://www.ohlsenwin.com/uploads/product-1.png)




[ N° CAS 5402-55-1 ] 2-thiophèneéthanol
norme de produit
| Apparence | Liquide transparent incolore à jaune clair |
| Test d'identité | Temps de rétention pour se conformer à la référence par GC |
| Pureté | 99 % min. (CG)) |
| 3-thiopnène éthanol | 0,1 % maximum (CG) |
| Impureté individuelle (inconnu) | 0,2 % maximum (CG) |
| Teneur en eau | 0,3 % maximum (W/W) |
| Période de retest | Un ans |
Détails du produit de [ 5402-55-1 ]
| N ° CAS. : | 5402-55-1 | N° MDL : | MFCD00005462 |
| Formule : | C6H8OS | Point d'ébullition : | 223°C à 760 mmHg |
| Formule de structure linéaire : | - | Clé InChI : | N / A |
| MW : | 128.19g/mole | Identifiant Pubchem : | 79400 |
| Synonymes : | |||
Chimie calculée de [ 5402-55-1 ]
Propriétés physicochimiques
| Num.atomes lourds : | 8 |
| Num.arom.atomes lourds : | 5 |
| Fraction Csp3 : | 0,33 |
| Num.liaisons rotatives : | 2 |
| Num.Accepteurs de liaison H : | 1.0 |
| Num.Donneurs de liaisons H : | 1.0 |
| Réfractivité molaire : | 35.25 |
| TSA : | 48.47Ų |
Pharmacocinétique
| Absorption gastro-intestinale : | Haut |
| Perméant BBB : | Oui |
| Substrat P-gp : | No |
| Inhibiteur du CYP1A2 : | No |
| Inhibiteur du CYP2C19 : | No |
| Inhibiteur du CYP2C9 : | No |
| Inhibiteur du CYP2D6 : | No |
| Inhibiteur du CYP3A4 : | No |
| Log Kp (perméation cutanée) : | -6,18 cm/s |
Lipophilie
| Log Po/w (iLOGP) : | 1,75 |
| Log Po/w (XLOGP3) : | 1.27 |
| Log Po/w (WLOGP) : | 1.28 |
| Log Po/w (MLOGP) : | 0,84 |
| Log Po/w (SILICOS-IT) : | 2,67 |
| Journal de consensus Po/w : | 1,56 |
Ressemblance à la drogue
| Lipinsky : | 0.0 |
| Ghose : | Aucun |
| Veber : | 0.0 |
| Egan : | 0.0 |
| Muegge : | 1.0 |
| Score de biodisponibilité : | 0,55 |
Solubilité dans l'eau
| Log S (ESOL) : | -1,77 |
| Solubilité : | 2,2 mg/ml ;0,0172 mol/l |
| Classe : | Très soluble |
| Journal S (Ali) : | -1,89 |
| Solubilité : | 1,66mg/ml ;0,013 mol/l |
| Classe : | Très soluble |
| Log S (SILICOS-IT) : | -1,86 |
| Solubilité : | 1,77mg/ml ;0,0138 mol/l |
| Classe : | Soluble |
Chimie médicale
| DES DOULEURS : | 0.0 alerte |
| Brenk : | 0.0 alerte |
| Ressemblance au plomb : | 1.0 |
| Accessibilité synthétique : | 1,82 |
Sécurité de [ 5402-55-1 ]
| Mot-indicateur : | Avertissement | Classe: | N / A |
| Conseils de prudence: | P261-P305+P351+P338 | ONU#: | N / A |
| Mentions de danger : | H302-H315-H319-H335 | Groupe d'emballage : | N / A |
| Pictogramme SGH : |  | ||
Conseils de prudence-Généralités
| Code | Phrase |
| P101 | Si un avis médical est nécessaire, ayez à portée de main le contenant ou l'étiquette du produit. |
| P102 | Tenir hors de portée des enfants. |
| P103 | Lire l'étiquette avant utilisation |
La prévention
| Code | Phrase |
| P201 | Obtenir des instructions spéciales avant utilisation. |
| P202 | Ne pas manipuler avant d'avoir lu et compris toutes les précautions de sécurité. |
| P210 | Tenir à l'écart de la chaleur/des étincelles/des flammes nues/des surfaces chaudes.- Interdiction de fumer. |
| P211 | Ne pas vaporiser sur une flamme nue ou sur une autre source d'inflammation. |
| P220 | Tenir/stocker à l'écart des vêtements/matériaux combustibles. |
| P221 | Prendre toutes les précautions pour éviter le mélange avec des combustibles |
| P222 | Ne pas laisser entrer en contact avec l'air. |
| P223 | Tenir à l'écart de tout contact possible avec de l'eau, en raison d'une réaction violente et d'un éventuel incendie éclair. |
| P230 | Garder mouillé |
| P231 | Manipuler sous gaz inerte. |
| P232 | Protéger de l'humidité. |
| P233 | Conserver le récipient bien fermé. |
| P234 | Conserver uniquement dans le contenant d'origine. |
| P235 | Garder son calme |
| P240 | Mettre à la terre/lier le conteneur et l'équipement de réception. |
| P241 | Utiliser des équipements électriques/de ventilation/d'éclairage/d'éclairage antidéflagrants. |
| P242 | Utilisez uniquement des outils anti-étincelles. |
| P243 | Prendre des mesures de précaution contre les décharges statiques. |
| P244 | Gardez les soupapes de réduction exemptes de graisse et d'huile. |
| P250 | Ne pas soumettre au meulage/choc/friction. |
| P251 | Récipient sous pression : Ne pas percer ni brûler, même après usage. |
| P260 | Ne pas respirer les poussières/fumées/gaz/brouillards/vapeurs/aérosols. |
| P261 | Éviter de respirer les poussières/fumées/gaz/brouillards/vapeurs/aérosols. |
| P262 | Ne pas mettre en contact avec les yeux, la peau ou les vêtements. |
| P263 | Éviter tout contact pendant la grossesse/pendant l'allaitement. |
| P264 | Bien se laver les mains après manipulation. |
| P265 | Bien laver la peau après manipulation. |
| P270 | Ne pas manger, boire ou fumer lors de l'utilisation de ce produit. |
| P271 | Utiliser uniquement à l'extérieur ou dans un endroit bien aéré. |
| P272 | Les vêtements de travail contaminés ne doivent pas sortir du lieu de travail. |
| P273 | Éviter le rejet dans l'environnement. |
| P280 | Porter des gants de protection/des vêtements de protection/un équipement de protection des yeux/du visage. |
| P281 | Utiliser l'équipement de protection individuelle requis. |
| P282 | Porter des gants isolants contre le froid/un écran facial/une protection oculaire. |
| P283 | Porter des vêtements résistants au feu/aux flammes/ignifuges. |
| P284 | Porter une protection respiratoire. |
| P285 | En cas de ventilation insuffisante, porter une protection respiratoire. |
| P231 + P232 | Manipuler sous gaz inerte.Protéger de l'humidité. |
| P235 + P410 | Garder son calme.Protéger du soleil. |
Réponse
| Code | Phrase |
| P301 | EN CAS D'INGESTION: |
| P304 | EN CAS D'INHALATION : |
| P305 | SI DANS LES YEUX : |
| P306 | SI SUR DES VÊTEMENTS : |
| P307 | SI exposé : |
| P308 | SI exposé ou concerné : |
| P309 | SI exposé ou si vous ne vous sentez pas bien : |
| P310 | Appeler immédiatement un CENTRE ANTIPOISON ou un médecin. |
| P311 | Appeler un CENTRE ANTIPOISON ou un médecin. |
| P312 | Appeler un CENTRE ANTIPOISON ou un médecin en cas de malaise. |
| P313 | Obtenir des conseils/des soins médicaux. |
| P314 | Consultez un médecin si vous ne vous sentez pas bien. |
| P315 | Obtenir immédiatement des conseils/des soins médicaux. |
| P320 | |
| P302 + P352 | EN CAS DE CONTACT AVEC LA PEAU : laver abondamment à l'eau et au savon. |
| P321 | |
| P322 | |
| P330 | Rincer la bouche. |
| P331 | NE PAS faire vomir. |
| P332 | EN CAS D'irritation DE LA PEAU : |
| P333 | En cas d'irritation ou d'éruption cutanée : |
| P334 | Immerger dans de l'eau froide/envelopper des bandages humides. |
| P335 | Brossez les particules libres de la peau. |
| P336 | Décongeler les parties givrées avec de l'eau tiède.Ne frottez pas la zone affectée. |
| P337 | Si l'irritation oculaire persiste : |
| P338 | Retirez les lentilles de contact, si elles sont présentes et faciles à faire.Continuez à rincer. |
| P340 | Transporter la victime à l'air frais et la maintenir au repos dans une position confortable pour respirer. |
| P341 | Si la respiration est difficile, amener la victime à l'air frais et la maintenir au repos dans une position confortable pour respirer. |
| P342 | En cas de symptômes respiratoires : |
| P350 | Laver délicatement avec beaucoup d'eau et de savon. |
| P351 | Rincer avec précaution à l'eau pendant plusieurs minutes. |
| P352 | Laver avec beaucoup de savon et d'eau. |
| P353 | Rincer la peau à l'eau/se doucher. |
| P360 | Rincer immédiatement les vêtements et la peau contaminés avec beaucoup d'eau avant de retirer les vêtements. |
| P361 | Retirer/Enlever immédiatement tous les vêtements contaminés. |
| P362 | Enlever les vêtements contaminés et les laver avant de les réutiliser. |
| P363 | Laver les vêtements contaminés avant de les réutiliser. |
| P370 | En cas d'incendie: |
| P371 | En cas d'incendie majeur et de grandes quantités : |
| P372 | Risque d'explosion en cas d'incendie. |
| P373 | NE PAS combattre le feu lorsque le feu atteint les explosifs. |
| P374 | Combattre l'incendie avec les précautions normales à une distance raisonnable. |
| P376 | Arrêtez la fuite si vous pouvez le faire en toute sécurité.Gaz comburants (section 2.4) 1 |
| P377 | Feu de fuite de gaz : Ne pas éteindre, sauf si la fuite peut être arrêtée en toute sécurité. |
| P378 | |
| P380 | Evacuer la zone. |
| P381 | Éliminer toutes les sources d'inflammation si cela est possible en toute sécurité. |
| P390 | Absorber le déversement pour éviter tout dommage matériel. |
| P391 | Recueillir le déversement.Dangereux pour le milieu aquatique |
| P301 + P310 | EN CAS D'INGESTION : Appeler immédiatement un CENTRE ANTIPOISON ou un médecin. |
| P301 + P312 | EN CAS D'INGESTION : appeler un CENTRE ANTIPOISON ou un médecin SI vous ne vous sentez pas bien. |
| P301 + P330 + P331 | EN CAS D'INGESTION : Rincer la bouche.NE PAS faire vomir. |
| P302 + P334 | EN CAS DE CONTACT AVEC LA PEAU : plonger dans de l'eau froide/envelopper dans des bandages humides. |
| P302 + P350 | EN CAS DE CONTACT AVEC LA PEAU : laver délicatement avec beaucoup d'eau et de savon. |
| P303 + P361 + P353 | EN CAS DE CONTACT AVEC LA PEAU (ou les cheveux) : Retirer/Enlever immédiatement tous les vêtements contaminés.Rincer la PEAU à l'eau/se doucher. |
| P304 + P312 | EN CAS D'INHALATION : Appeler un CENTRE ANTIPOISON ou un médecin si vous ne vous sentez pas bien. |
| P304 + P340 | EN CAS D'INHALATION : Transporter la victime à l'air frais et la maintenir au repos dans une position où elle peut confortablement respirer. |
| P304 + P341 | EN CAS D'INHALATION : Si la respiration est difficile, amener la victime à l'air frais et la maintenir au repos dans une position confortable pour respirer. |
| P305 + P351 + P338 | EN CAS DE CONTACT AVEC LES YEUX : rincer avec précaution à l'eau pendant plusieurs minutes.Retirez les lentilles de contact, si elles sont présentes et faciles à faire.Continuez à rincer. |
| P306 + P360 | EN CAS DE VÊTEMENTS : Rincer immédiatement les VÊTEMENTS et la PEAU contaminés avec beaucoup d'eau avant de retirer les vêtements. |
| P307 + P311 | EN CAS d'exposition : appeler un CENTRE ANTIPOISON ou un médecin. |
| P308 + P313 | SI exposé ou concerné : consulter un médecin. |
| P309 + P311 | EN CAS d'exposition ou de malaise : appeler un CENTRE ANTIPOISON ou un médecin. |
| P332 + P313 | EN CAS D'irritation DE LA PEAU : Consulter un médecin. |
| P333 + P313 | EN CAS D'irritation ou d'éruption cutanée : Consulter un médecin. |
| P335 + P334 | Brossez les particules libres de la peau.Plonger dans de l'eau froide/envelopper dans des bandages humides. |
| P337 + P313 | SI l'irritation oculaire persiste : Consulter un médecin. |
| P342 + P311 | EN CAS de symptômes respiratoires : appeler un CENTRE ANTIPOISON ou un médecin. |
| P370 + P376 | En cas d'incendie : Arrêter la fuite si cela peut être fait sans danger. |
| P370 + P378 | En cas d'incendie: |
| P370 + P380 | En cas d'incendie : Evacuer la zone. |
| P370 + P380 + P375 | En cas d'incendie : Evacuer la zone.Combattre l'incendie à distance en raison du risque d'explosion. |
| P371 + P380 + P375 | En cas d'incendie majeur et de grandes quantités : Evacuer la zone.Combattre l'incendie à distance en raison du risque d'explosion. |
Stockage
| Code | Phrase |
| P401 | |
| P402 | Entreposer dans un endroit sec. |
| P403 | Conserver dans un endroit bien aéré. |
| P404 | Conserver dans un contenant fermé. |
| P405 | Magasin fermé à clé. |
| P406 | Stocker dans un récipient résistant à la corrosion/avec une doublure intérieure résistante. |
| P407 | Maintenir un espace d'air entre les piles/palettes. |
| P410 | Protéger du soleil. |
| P411 | |
| P412 | Ne pas exposer à des températures supérieures à 50 oC/ 122 oF. |
| P413 | |
| P420 | Stocker à l'écart d'autres matériaux. |
| P422 | |
| P402 + P404 | Entreposer dans un endroit sec.Conserver dans un contenant fermé. |
| P403 + P233 | Conserver dans un endroit bien aéré.Conserver le récipient bien fermé. |
| P403 + P235 | Conserver dans un endroit bien aéré.Garder son calme. |
| P410 + P403 | Protéger du soleil.Conserver dans un endroit bien aéré. |
| P410 + P412 | Protéger du soleil.Ne pas exposer à des températures supérieures à 50 oC/122 oF. |
| P411 + P235 | Garder son calme. |
Disposition
| Code | Phrase |
| P501 | Éliminer le contenu/récipient dans ... |
| P502 | Se référer au fabricant/fournisseur pour des informations sur la récupération/recyclage |
Dangers physiques
| Code | Phrase |
| H200 | Explosif instable |
| H201 | Explosif;risque d'explosion en masse |
| H202 | Explosif;risque de projection important |
| H203 | Explosif;risque d'incendie, d'explosion ou de projection |
| H204 | Risque d'incendie ou de projection |
| H205 | Peut exploser en masse dans le feu |
| H220 | Gaz extrêmement inflammable |
| H221 | Gaz inflammable |
| H222 | Aérosol extrêmement inflammable |
| H223 | Aérosol inflammable |
| H224 | Liquide et vapeur extrêmement inflammables |
| H225 | Liquide et vapeur très inflammables |
| H226 | Liquide et vapeur inflammables |
| H227 | Liquide combustible |
| H228 | Solide inflammable |
| H229 | Récipient sous pression : peut éclater s'il est chauffé |
| H230 | Peut réagir de manière explosive même en l'absence d'air |
| H231 | Peut réagir de manière explosive même en l'absence d'air à une pression et/ou une température élevées |
| H240 | Le chauffage peut provoquer une explosion |
| H241 | Le chauffage peut provoquer un incendie ou une explosion |
| H242 | Le chauffage peut provoquer un incendie |
| H250 | S'enflamme spontanément si exposé à l'air |
| H251 | Auto-échauffement ;peut prendre feu |
| H252 | Auto-échauffement en grande quantité;peut prendre feu |
| H260 | Au contact de l'eau, dégage des gaz inflammables qui peuvent s'enflammer spontanément |
| H261 | Au contact de l'eau dégage des gaz inflammables |
| H270 | Peut provoquer ou aggraver un incendie ;comburant |
| H271 | Peut provoquer un incendie ou une explosion ;oxydant fort |
| H272 | Peut intensifier le feu ;comburant |
| H280 | Contient du gaz sous pression;peut exploser si chauffé |
| H281 | Contient du gaz réfrigéré ;peut causer des brûlures cryogéniques ou des blessures |
| H290 | Peut être corrosif pour les métaux |
Dangers pour la santé
| Code | Phrase |
| H300 | Mortel en cas d'ingestion |
| H301 | Toxique en cas d'ingestion |
| H302 | Nocif en cas d'ingestion |
| H303 | Peut être nocif en cas d'ingestion |
| H304 | Peut être mortel en cas d'ingestion et de pénétration dans les voies respiratoires |
| H305 | Peut être nocif en cas d'ingestion et de pénétration dans les voies respiratoires |
| H310 | Mortel par contact avec la peau |
| H311 | Toxique au contact de la peau |
| H312 | Nocif par contact avec la peau |
| H313 | Peut être nocif au contact de la peau |
| H314 | Provoque de graves brûlures de la peau et des lésions oculaires |
| H315 | Provoque une irritation de la peau |
| H316 | Provoque une légère irritation de la peau |
| H317 | Peut provoquer une réaction allergique cutanée |
| H318 | Provoque de graves lésions oculaires |
| H319 | Provoque une grave irritation des yeux |
| H320 | Provoque une irritation des yeux |
| H330 | Mortel si inhalé |
| H331 | Toxique si inhalé |
| H332 | Nocif en cas d'inhalation |
| H333 | Peut être nocif en cas d'inhalation |
| H334 | Peut provoquer des symptômes d'allergie ou d'asthme ou des difficultés respiratoires en cas d'inhalation |
| H335 | Peut provoquer une irritation des voies respiratoires |
| H336 | Peut causer de la somnolence ou des étourdissements |
| H340 | Peut causer des défauts génétiques |
| H341 | Susceptible de provoquer des anomalies génétiques |
| H350 | Peut causer le cancer |
| H351 | Susceptible de provoquer le cancer |
| H360 | Peut nuire à la fertilité ou à l'enfant à naître |
| H361 | Susceptible de nuire à la fertilité ou à l'enfant à naître |
| H361d | Susceptible de nuire à l'enfant à naître |
| H362 | Peut être nocif pour les enfants allaités |
| H370 | Cause des dommages aux organes |
| H371 | Peut causer des dommages aux organes |
| H372 | Cause des dommages aux organes par exposition prolongée ou répétée |
| H373 | Peut causer des dommages aux organes en cas d'exposition prolongée ou répétée |
Dangers environnementaux
| Code | Phrase |
| H400 | Très toxique pour la vie aquatique |
| H401 | Toxique pour la vie aquatique |
| H402 | Nocif pour la vie aquatique |
| H410 | Très toxique pour la vie aquatique avec effets à long terme |
| H411 | Toxique pour la vie aquatique avec des effets durables |
| H412 | Nocif pour la vie aquatique avec des effets durables |
| H413 | Peut causer des effets nocifs à long terme sur la vie aquatique |
| H420 | Nuis à la santé publique et à l'environnement en détruisant l'ozone dans la haute atmosphère |
Demande en synthèse de [ 5402-55-1 ]
Voie de synthèse amont du [ 5402-55-1 ]
Voie de synthèse aval du [ 5402-55-1 ]
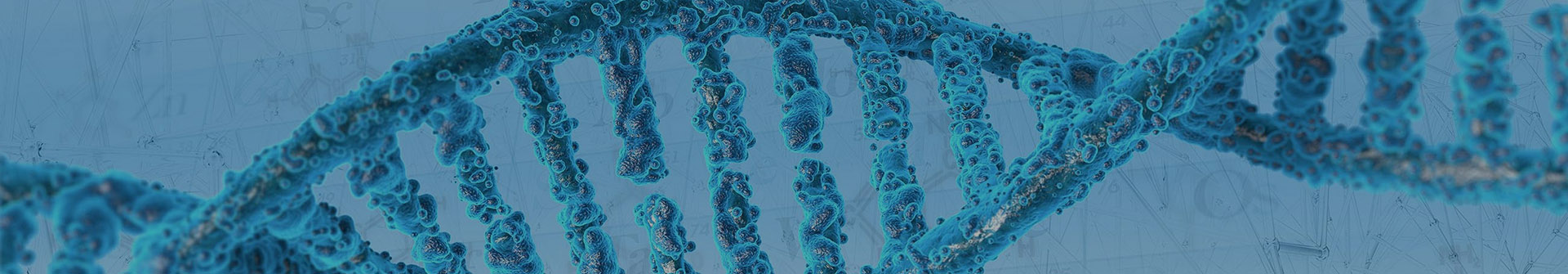
Voie de synthèse amont du [ 5402-55-1 ]

| Rendement | Conditions de réaction | Fonctionnement en expérience |
|---|---|---|
| 72% | Avec la souche du champignon zygomycète S.racemosum MUT 770 Dans du diméthylsulfoxyde pendant 72 h ;Réaction enzymatique | 2.3.Expériences de biotransformation Des souches fongiques ont été pré-cultivées dans des boîtes de Pétri contenant un milieu solide d'extrait de malt (MEA : 20 g L-1 de glucose, 20 g L-1 d'extrait de malt, 20 g L-1 d'agar, 2 g L-1 de peptone) à partir de laquelle l'inoculum pour les cultures liquides a été mis en place.Le champignon a été inoculé sous forme de suspension de conidies (1 106 conidies/mL) dans des asks de 50 mL contenant 40 mL de milieu liquide extrait de malte.Les flacons ont été incubés à 25 °C et maintenus sous agitation (110 tr/min). Après 2 jours de pré-croissance, une solution à 500 mM du substrat dans le DMSO a été ajoutée, à une concentration de substrat de départ (c0) de 1 à 5 mM.Pour chaque substrat, trois répétitions biologiques ont été exécutées. L'expérience a duré 3 jours après l'ajout des substrats, période pendant laquelle des échantillons de 1 ml ont été prélevés, à des intervalles spécifiés (généralement 24, 48 et 72 h).Chaque échantillon a été extrait avec EtOAc (500 L), la phase organique a été séchée sur Na2S04 anhydre et analysée au moyen de GC/MS.Dans certains cas (voir section 2.4) l'isolement du produit réduit a été réalisé. Pour chaque ensemble de biotransformations, une demande a été utilisée pour mesurer la biomasse et le pH initiaux avant l'ajout du substrat. Ces paramètres ont également été évalué à la fin de l'expérience pour toutes les demandes.Le milieu liquide a été séparé de la biomasse par filtration et a été utilisé pour la mesure du pH tandis que les mycéliums ont été séchés à 60°C pendant 24 h pour mesurer le poids sec de la biomasse.2-(thiophène-2-yl)éthanol : à partir d'acide 2-(thiophén-2-yl)acétique (3,7 mg, 72 %) et de 2-(thiophén-2-yl)acétate de méthyle (24,6 mg, 96 %).RMN 1H (400 MHz, CDCl3, TMS) : = 7,20 (m, 1H, hydrogène hétéroaromatique), 6,99 (m, 1H, hydrogène hétéroaromatique), 6,90 (m,1H, hydrogène hétéroaromatique), 3,85 (t, 2H, J = 6,2 Hz, CH2OH), 3,02(t, 2H, J = 6,2 Hz, CH2CH2OH).RMN 13C (100 MHz, CDC13, TMS) : = 140,5, 127,0, 125,8, 124,0, 63,4, 33,3.GC/MS : tR = 9,47 min, m/z128 (M+, 30), 110 (5), 97 (100) |
| 66,5 % | Étape #1 : Avec le tétrahydrure d'aluminium de lithium dans le tétrahydrofurane à 0 - 20℃ ; Étape #2 : Avec de l'eau ;chlorure de sodium dans le tétrahydrofuranne à 0℃ ; | Étape 9 2-(thiophén-2-yl)éthanol : Vers 0°C, une solution d'acide thiophén-2-yl-acétique (1,0 g ; 7,03 mmol) dans le tétrahydrofuranne (10 mL) a été ajoutée goutte à goutte à une suspension d'hydrure de lithium et d'aluminium (0,534 g ; 14,05 mmol) dans du liquide sec. tétrahydrofuranne (10 ml). Le mélange a été agité à température ambiante pendant environ 4 heures, puis refroidi à environ 0°C. Après addition d'une solution saturée froide de chlorure de sodium (1 ml), le mélange a été filtré et les sels inorganiques ont été lavés avec du tétrahydrofurane et de l'acétate d'éthyle. Le filtrat et les lavages ont été combinés et concentrés sous vide pour donner le composé du titre sous forme d'huile brune (0,600 g; 66,5 pour cent). 1RMN H (400 MHz, CDCl3) δ 1,60 (br, échangeable avec D2O, 1H), 3,08 (t, J = 6,2 Hz, 2H), 3,85 (t, J = 6,2 Hz, 2H), 6,87-6,88 (m, 1H), 6,95-6,97 (m, 1H), 7,16-7,25 (m, 1H).IR (film) υ 3345, 3105, 2211, 2126, 2090, 1792, 1433, 1138, 972, 737, 699 cm-1MS : 129 (M+1). |

| Rendement | Conditions de réaction | Fonctionnement en expérience |
|---|---|---|
| 96% | Avec la souche du champignon zygomycète S.racemosum MUT 770 Dans du diméthylsulfoxyde pendant 72 h ;Réaction enzymatique | 2.3.Expériences de biotransformation Des souches fongiques ont été pré-cultivées dans des boîtes de Pétri contenant un milieu solide d'extrait de malt (MEA : 20 g L-1 de glucose, 20 g L-1 d'extrait de malt, 20 g L-1 d'agar, 2 g L-1 de peptone) à partir de laquelle l'inoculum pour les cultures liquides a été mis en place.Le champignon a été inoculé sous forme de suspension de conidies (1 106 conidies/mL) dans des asks de 50 mL contenant 40 mL de milieu liquide extrait de malte.Les flacons ont été incubés à 25 °C et maintenus sous agitation (110 tr/min). Après 2 jours de pré-croissance, une solution à 500 mM du substrat dans le DMSO a été ajoutée, à une concentration de substrat de départ (c0) de 1 à 5 mM.Pour chaque substrat, trois répétitions biologiques ont été exécutées. L'expérience a duré 3 jours après l'ajout des substrats, période pendant laquelle des échantillons de 1 ml ont été prélevés, à des intervalles spécifiés (généralement 24, 48 et 72 h).Chaque échantillon a été extrait avec EtOAc (500 L), la phase organique a été séchée sur Na2S04 anhydre et analysée au moyen de GC/MS.Dans certains cas (voir section 2.4) l'isolement du produit réduit a été réalisé. Pour chaque ensemble de biotransformations, une demande a été utilisée pour mesurer la biomasse et le pH initiaux avant l'ajout du substrat. Ces paramètres ont également été évalué à la fin de l'expérience pour toutes les demandes.Le milieu liquide a été séparé de la biomasse par filtration et a été utilisé pour la mesure du pH tandis que les mycéliums ont été séchés à 60°C pendant 24 h pour mesurer le poids sec de la biomasse.2-(thiophène-2-yl)éthanol : à partir d'acide 2-(thiophén-2-yl)acétique (3,7 mg, 72 %) et de 2-(thiophén-2-yl)acétate de méthyle (24,6 mg, 96 %).RMN 1H (400 MHz, CDCl3, TMS) : = 7,20 (m, 1H, hydrogène hétéroaromatique), 6,99 (m, 1H, hydrogène hétéroaromatique), 6,90 (m,1H, hydrogène hétéroaromatique), 3,85 (t, 2H, J = 6,2 Hz, CH2OH), 3,02(t, 2H, J = 6,2 Hz, CH2CH2OH).RMN 13C (100 MHz, CDC13, TMS) : = 140,5, 127,0, 125,8, 124,0, 63,4, 33,3.GC/MS : tR = 9,47 min, m/z128 (M+, 30), 110 (5), 97 (100) |
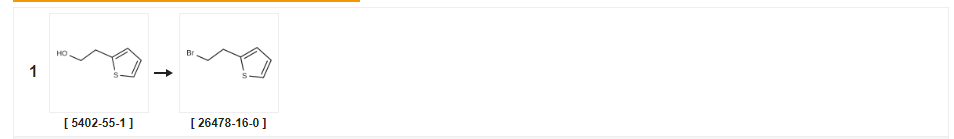
Voie de synthèse aval du [ 5402-55-1 ]
| Rendement | Conditions de réaction | Fonctionnement en expérience |
|---|---|---|
| 94% | Avec du tétrabromure de carbone;triphénylphosphine dans le tétrahydrofurane à 0℃ ;Technique de Schlenk;Atmosphère inerte ; | |
| 69% | Avec du brome;triphénylphosphine dans du dichlorométhane à 20℃ ; | |
| 64% | P.31 Synthèse du 2-(2-bromoéthyl)thiophène Exemple de production 31 Synthèse de 2-(2-bromoéthyl)thiophène Du 2-thiényléthanol (0,44 ml) a été traité comme dans l'exemple de production 1 ci-dessus pour donner le composé du titre (0,490 g) sous la forme d'une huile incolore (rendement : 64,0 %).1H-RMN (400 MHz, CDCl3) : δ(ppm) 3,38(2H, t, J=7,6Hz), 3,58(2H, t, J=7,6Hz), 6,89(1H, d, J=1,2Hz), 6,96 (1H, d, J = 4,2 Hz), 7,19 (1H, dd, J = 1,2, 4,2 Hz). |
| 28% | Avec du tribromure de phosphore dans du dichlorométhane à 0 - 20℃ ;pendant 1h ; | |
| 19% | Avec du tribromure de phosphore dans du tétrachlorométhane à 65℃ ;pour 0,333333h ; | a Du tribromure de phosphore (2,0 mL, 21,1 mmol) a été ajouté à une solution agitée de 2-(thiophén-2-yl)éthanol (2,25 g, 17,6 mmol) dans du tétrachlorure de carbone (162 mL) puis le mélange a été chauffé à 65°C pendant 20 minutes.Le mélange a été laissé refroidir à température ambiante puis de la glace a été ajoutée.La couche organique a été séparée puis la couche aqueuse a été extraite avec du dichlorométhane (2 x 30 mL).Les couches organiques combinées ont été lavées avec de la saumure, puis séchées (NaS04), filtrées et réduites sous vide.Le résidu a été purifié par Chromatographie éclair sur de la silice, en éluant avec des mélanges acétate d'éthyle:hexane 0:100 à 0,5:95,5 pour donner le 2-(2-bromoéthyl)thiophène sous la forme d'une huile brune (650 mg, 19%). |
| Avec de la pyridine ;chloroforme;tribromure de phosphore | ||
| Avec du tribromure de phosphore dans de l'éther diéthylique à 0℃ ;pendant 4h; | 2D Exemple 2D ;9-(4,5-Diméthyl-thiazol-2-yl)-4-[4-(2-thiophén-2-yl-éthyl)-pipérazin-1-yl]-5,6,7, 8- TETRAHYDRO- 1, 3, 4B-TRIAZA-FLUORÈNE A une solution de 2-(2-thiényl)éthanol (1,63 g) dans de l'éther sec (15 ml) à 0°C, on a ajouté goutte à goutte du PBr3 (1,31 ml).Après 4 heures, le mélange réactionnel a été dilué avec du dichlorométhane, lavé avec de l'eau, séché (MGS04) et le solvant éliminé sous vide pour donner une huile brune qui a été purifiée par chromatographie flash pour donner du 2-(2-bromo-éthyl)-thiophène.Un mélange de 9-(4,5-DIMETHYL-THIAZOL-2-YL)-4-PIPERAZIN-1-YL-5,6,7,8-TETRAHYDRO- 1,3,4b-triaza-fluorène (85mg), Du 2-(2-bromo-éthyl)-thiophène (44 mg) et du carbonate de potassium (38 mg) ont été chauffés au reflux dans de l'acétonitrile (5 ml) pendant 4 heures.Le mélange réactionnel a été refroidi, extrait dans du dichlorométhane, séché (MGS04) et le solvant a été éliminé sous vide pour donner le produit brut qui a été purifié par Chromatographie éclair pour donner le composé du titre (30 mg). |
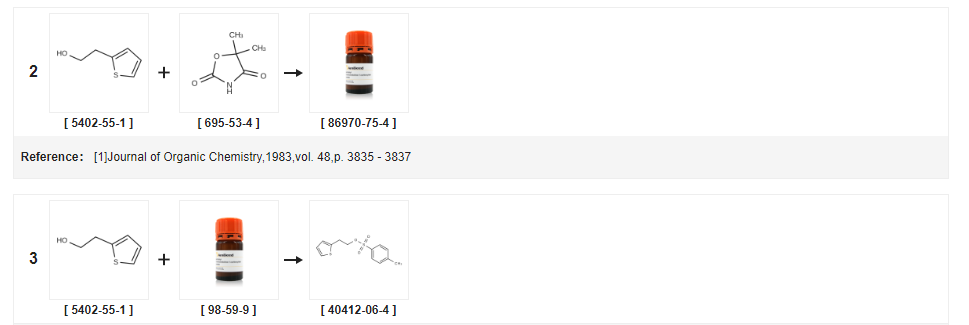
| Rendement | Conditions de réaction | Fonctionnement en expérience |
|---|---|---|
| 98% | Avec triéthylamine;à 35 ℃ ; Refroidissement avec de la glace ; | (0,87 mol) de 2-thiophèneéthanol et 184 g (0,97 mol) de chlorure de p-toluènesulfonyle ont été ajoutés séquentiellement dans un ballon tricol de 1 L, 98 g (0,97 mol) de triéthylamine y ont été ajoutés goutte à goutte sous un bain d'eau glacée, Et maintenir la température du liquide de réaction non supérieure à 20 degrés C, laisser tomber, chauffer à 35 degrés C pour continuer à remuer, respectivement 24h, 27h d'échantillonnage, TLC, jusqu'à ce que la réaction soit terminée, arrêter la réaction, la filtration, filtrer le gâteau avec le produit approprié quantité de dichlorométhane Et la couche de chlorure de méthylène a été séchée avec du sulfate de sodium anhydre pendant 2 heures.Le déshydratant a été séparé par filtration et le déshydratant a été lavé avec une petite quantité de chlorure de méthylène.Le filtrat a été décomprimé sous vide et le filtrat a été évaporé sous pression réduite.Concentré à poids constant pour être une huile brune, pesant 203g, rendement 98%, |
| 96,37 % | Avec triéthylamine;Dans le toluène ;à 5 - 30℃ ;pour 20.8333h ; Distribution / sélectivité des produits ; | EXEMPLE 3 Préparation de para-toluènesulfonate de 2-thiényléthyle (formule VII) à l'aide de toluène Du 2-éthanol a été ajouté à environ 5°C en environ 20 minutes suivi de l'addition de 130 kg de triéthylamine en environ 8 heures 50 minutes.La température du mélange réactionnel a été portée à environ 30°C puis agitée pendant environ 12 heures.La masse réactionnelle a été filtrée sur un filtre Nutsche et lavée avec 2 x 100 litres de toluène.Le filtrat réactionnel a été transféré dans un autre réacteur suivi d'un lavage avec 5 x 200 litres d'eau.Les couches organique et aqueuse ont été séparées et la couche organique a été distillée complètement à environ moins de 70°C sous vide pour donner 212 kg (rendement : 96,37 %) du composé du titre.Pureté par GC : 95,59 %. |
| 96,5 % | Avec triéthylamine;Dans le dichlorométhane ;à -5 - 20℃ ;pendant 2h; | 32,7 g (0,17 mol) de chlorure de p-toluènesulfonyle, 40 ml de dichlorométhane dans le ballon de réaction, refroidi à -5 C, 20 g (0,16 mol) de 2-thiophèneéthanol. 28,4 g (0,28 mol) de triéthylamine ont été lentement ajoutés goutte à goutte et la température de la solution réactionnelle a été maintenue à environ 0 C. Plus Bi, incubé pendant 2 h après que la réaction a été réchauffée à température ambiante.2-thiophène éthanol à consommer jusqu'aux matières premières, filtration par aspiration, Le solide a été lavé avec une petite quantité de chlorure de méthylène et le filtrat a été lavé avec 50 ml de bicarbonate de sodium saturé et séché sur sulfate de sodium anhydre.Filtration, concentration du filtrat, solide brun clair précipité, filtré, lavé avec une petite quantité d'éther de pétrole à blanc, c'est-à-dire ester éthylique de P-toluènesulfonate-2-thiophène 42,5 g, rendement 96,5 % (pureté HPLC 99 %) |
| 95,5 % | Avec triéthylamine;Dans le dichlorométhane ;à 7,5 - 22,5℃ ;pendant 5h ; Distribution / sélectivité des produits ; | EXEMPLE 10 Préparation de tosylate de 2-(2-thiophène)éthanol (Formule VII) à l'aide de dichlorométhane Dans un réacteur à une température d'environ 30 C, on a ajouté 4 litres de dichlorométhane, refroidi à une température d'environ 7,5 C, puis on ajoute 1,784 kg de chlorure de p-toluène sulfonyle suivi de 1 kg de thiophène-2-éthanol.1,302 kg de triéthylamine ont été ajoutés à la masse réactionnelle ci-dessus à une température d'environ 7,5°C suivi d'une élévation lente de la température de la masse réactionnelle à 22,5°C pendant environ 5 heures.La masse réactionnelle obtenue a été filtrée à travers un filtre Nutsche sous pression, lavée avec du chlorure de méthylène (2 x 1 litre) et la liqueur mère a été recueillie et transférée dans un autre réacteur.La couche organique a été lavée avec de l'eau (5 x 2 litres).La couche organique ainsi obtenue a été soumise à une distillation à une température inférieure à 70°C en utilisant une circulation d'eau chaude.Le résidu obtenu a ensuite été refroidi à environ 30°C pour donner 2,1 kg (rendement : 95,5 %) du composé du titre. |
| 93,6 % | Avec triéthylamine;Dans le toluène ;à 45℃ ;pendant 4h; | Exemple 1 0.2.Conversion du bromhydrate de (S)-1,2,3,4-tétrahydro-5-hvdroxy-N-propyl-naphtalène-2-ammonium (VIII) en chlorhydrate de rotigotine;10.2.1.Préparation de sulfonate de 2-(2-thiényl)éthyl-4-toluène;Le chlorure de 4-toluènesulfonyle (162 g), le toluène (363,3 g) et le 2-(2-thiényl)éthanol (104 g) sont combinés.De la triéthylamine (93 g) est ajoutée en maintenant la température inférieure à 45°C. Après 4h, le mélange est lavé avec de l'acide phosphorique aqueux, de la soude aqueuse puis de l'eau.La phase organique est distillée sous vide.De l'isopropanol (314 g) et des heptanes (365,9 g) sont ajoutés.Le lot est cristallisé par refroidissement et isolé à -15 C. Les cristaux sont filtrés et lavés avec des heptanes (175 mL).Les cristaux sont ensuite séchés sous vide à température ambiante jusqu'à l'obtention d'un point de fusion > 30 C. Rendement ( 214 g) : 93,6 % Les analyses HPLC ont confirmé une pureté > 99 % et un dosage de 100 % par rapport à un étalon de référence. |
| 93,6 % | Avec triéthylamine;Dans le toluène ;à 45℃ ;pendant 4h; | Le chlorure de 4-toluènesulfonyle (162 g), le toluène (363,3 g) et le 2-(2-thiényl)éthanol (104 g) sont combinés.De la triéthylamine (93 g) est ajoutée en maintenant la température inférieure à 45 C. Après 4 h, le mélange est lavé avec de l'acide phosphorique aqueux, de la soude aqueuse puis de l'eau.La phase organique est distillée sous vide.De l'isopropanol (314 g) et des heptanes (365,9 g) sont ajoutés.Le lot est cristallisé par refroidissement et isolé à -15 C. Les cristaux sont filtrés et lavés avec des heptanes (175 mL).Les cristaux sont ensuite séchés sous vide à température ambiante jusqu'à l'obtention d'un point de fusion de 30°C.Rendement (214 g) : 93,6 % |
| 90% | Avec gel de silice;Dans le dichlorométhane ;pendant 2h ; Reflux ; | Les 12,8 g (0,1 mol) de 2- (2- thiényl) éthanol, 1000 ml de dichlorométhane, 21,0 g (0,12 mol) de chlorure d'acide p-toluènesulfonique et 10,0 g de gel de silice dans le ballon de réaction, la réaction a été chauffée au reflux pendant 2 h, refroidi, filtré pour éliminer le gel de silice.Le mélange réactionnel a été lavé successivement avec de l'eau distillée, une solution saturée de carbonate de sodium, de la saumure, puis le solvant chlorure de méthylène a été éliminé par distillation sous pression réduite, pour donner 26,0g d'acide p-toluènesulfonique Préparation de 2 - Ester de (2-thiényl)éthyle, rendement 90 %. |
| Dans la pyridine ;eau; | (a) Tosylate de 2-(2-hydroxyéthyl)thiophène Du chlorure de toluène-4-sulfonyle (4,125 g) a été ajouté par portions en 5 minutes à une solution glacée de 2-(2-hydroxyéthyl)thiophène (1,723 g) dans de la pyridine anhydre (20 ml) et la solution jaune pâle résultante a été agitée à 0°C. Au bout de 3 h, le mélange réactionnel a été versé dans de l'eau vigoureusement agitée (160 ml) produisant un précipité.Après refroidissement, le solide a été recueilli et lavé avec de l'eau pour donner des cristaux blancs du tosylate du titre (3,555 g), mp 33-34°C, lambdamax (EtOH) 227 nm (E11 612). | |
| Avec triéthylamine;Dans la butanone ;à 0 - 30℃ ; | Exemple 1 Préparation de para-toluènesulfonate de 2-(2-thiényl)-éthyl Un mélange de chlorure de p-toluènesulfonyle (328 g) et de 2-(2-thiényl)-éthanol (200 g) dans de la méthyléthylcétone (1000 ml) a été refroidi à 0C.Cela a été suivi par l'addition goutte à goutte de triéthylamine (283,1 ml) à 0-5°C sur une période de 1 à 2 heures, et la masse réactionnelle a été agitée pendant 12 à 15 heures à 25-30°C.La masse résultante a été filtrée et lavée avec de la méthyléthylcétone (500 ml).La couche organique résultante a été lavée avec de l'eau (500 ml) suivi de lavages avec une solution saturée de bicarbonate de sodium (500 ml) et une solution de saumure (500 ml).La couche organique résultante a été distillée sous vide à moins de 50 °C pour donner du para-toluènesulfonate de 2-(2-thiényl)-éthyle sous la forme d'une masse huileuse (poids de l'huile : 505 g ; pureté par HPLC : 97 %). | |
| Avec triéthylamine;Dans la butanone ;à 0 - 30℃ ; | Exemple 1 Préparation de para-toluènesulfonate de 2-(2-thiényl)-éthyl Un mélange de chlorure de p-toluènesulfonyle (328 g) et de 2-(2-thiényl)-éthanol (200 g) dans de la méthyléthylcétone (1000 ml) a été refroidi à 0°C. Cela a été suivi par l'addition goutte à goutte de triéthylamine (283,1 ml) à 0-5°C sur une période de 1 à 2 heures, et la masse réactionnelle a été agitée pendant 12 à 15 heures à 25-30°C. La masse résultante a été filtrée et lavée avec de la méthyléthylcétone (500 ml).La couche organique résultante a été lavée avec de l'eau (500 ml) suivi de lavages avec une solution saturée de bicarbonate de sodium (500 ml) et une solution de saumure (500 ml).La couche organique résultante a été distillée sous vide à moins de 50°C pour donner du para-toluènesulfonate de 2-(2-thiényl)-éthyle sous la forme d'une masse huileuse (Poids de l'huile : 505 g ; Pureté par HPLC : 97 %). | |
| Avec du carbonate de potassium ;Dans le toluène ;à 0 - 20℃ ;pendant 3h30 ; | 500 ml de toluène ont été ajoutés à un ballon de réaction de 1000 ml, 50 g d'éthanol de thiophène et 80 g de chlorure de p-toluène sulfonyle, Allumer l'agitation, Une température contrôlée a été ajoutée goutte à goutte à 0 C64 g de carbonate de potassium, L'addition a été poursuivie pendant environ 30 min, Chute terminé le réchauffement à 20 C de réaction pendant 3 heures,À la solution de réaction a été ajouté 400 ml d'eau,Lavage deux fois,La couche de toluène lavée a été directement utilisée pour la réaction suivante. | |
| Avec la N-éthyl-N,N-diisopropylamine;Dans le toluène ;à 0 - 20℃ ;pendant 3h30 ; | 500 ml de toluène ont été ajoutés dans un ballon de réaction de 1000 ml, 50 g de thiophène éthanol et 92 g de chlorure de p-toluènesulfonyle, agités à 0 C, 62 g de N,N-diisopropyléthylamine ont été ajoutés goutte à goutte pendant environ 30min.Une fois le goutte-à-goutte terminé, le mélange a été chauffé à 20°C et mis à réagir pendant 3 heures.A la solution réactionnelle ont été ajoutés 400 ml d'eau, lavés deux fois, la couche de toluène lavée a été directement utilisée pour la réaction suivante. | |
| Avec triéthylamine;Dans le toluène ;à 0 - 20℃ ;pendant 3h; | 500 ml de toluène, 50 g d'éthanol thiophène et 80 g de chlorure de p-toluènesulfonyle ont été placés dans un ballon de réaction de 1000 ml, et la température a été portée à 20 C pendant 3 heures.400 ml d'eau ont été ajoutés à la solution réactionnelle, lavés deux fois, lavés avec du toluène, couche directement à la réaction de l'étape suivante. | |
| Avec du carbonate de potassium ;Dans le toluène ;à 0 - 20℃ ;pendant 3h30 ; | 500 ml de toluène, 50 g d'éthanol de thiophène et 107 g de chlorure de p-toluènesulfonyle ont été placés dans un ballon de réaction de 1000 ml, l'agitation a été activée, la température a été contrôlée à 0 C, addition goutte à goutte de 64 g de carbonate de potassium, l'addition a duré environ 30 minutes, après quoi il a été réchauffé à 20°C et mis à réagir pendant 3 heures, à la solution réactionnelle ont été ajoutés 400 ml d'eau, lavés deux fois, et la couche de toluène lavée a été directement utilisée pour la réaction suivante. | |
| Avec triéthylamine;Dans le toluène ;à 0 - 25℃ ;pendant 3h30 ; | Exemple 1 Le procédé de préparation du chlorhydrate de ticlopidine tel que représenté sur la figure 2 comprend les étapes suivantes :1.Protection du p-toluènesulfonyle : Dans un ballon réactionnel de 1000 ml, 500 ml de toluène, 50 g d'éthanol de thiophène et 80 g de chlorure de p-toluènesulfonyle ont été chargés et agités pour contrôler. A une température de 0 C, 47 g de triéthylamine ont été ajoutés goutte à goutte, goutte pendant environ 30 min, et la température a été portée à 25 C pendant 3 heures.Ajouter 400 ml d'eau, lavé deux fois, lavé avec une couche de toluène directement à la réaction de l'étape suivante.2.Réaction de condensation La solution réactionnelle de toluène de l'étape précédente a été ajoutée à un ballon de réaction de 1000 ml, suivi par l'addition de 114 g d'o-chlorobenzylamine et le chauffage à 90°C pendant 3 heures.Après la réaction, le mélange a été refroidi à 25 C et agité pendant 1 heure.Le filtrat a été additionné de 200 ml d'eau et d'acide chlorhydriqueAjuster le pH du système à 8,5, stratifier la couche supérieure de toluène et continuer à faire goutter de l'acide chlorhydrique pour ajuster le pH à 5, puis refroidir le système à 2 CLes cristaux ont été mélangés pendant 4 heures et filtré, et le gâteau de filtration a été séché sous vide à 50°C pour donner 96 g du chlorhydrate de condensat.3 ?Réaction en boucle fermée Au ballon de réaction de 1000 ml, on a ajouté 96 g du chlorhydrate du condensat, 400 ml de 1,3-dioxane et 5 ml d'acide chlorhydrique, à 90°C pendant 6 heures.Après la réaction, le mélange a été refroidi à 7°C et agité pendant 3 heures.Le gâteau de filtration a été lavé avec une petite quantité de 1,3-dioxétane. Après séchage sous vide à 50°C, 95 g de chlorhydrate de ticlopidine ont été obtenus4 ?RaffinéDans un flacon de réaction de 1000 ml, 95 g de chlorhydrate de ticlopidine brut et 500 ml d'éthanol absolu ont été ajoutés et chauffés à 72 C sous agitation, environ 10 minutes après la dissolution complète du solide en ajoutant 2 g de charbon actif, le blanchiment de l'isolation 20 minutes après le filtre chaud, le filtrat refroidit progressivement. Les cristaux sont cristallisés à 4 C pendant 4 heures et filtrés.Le gâteau de filtre a été lavé avec une petite quantité d'éthanol absolu et séché sous vide à 50°C pour donner 91 g de produits de précision de chlorhydrate de tithiol.Rendement total 82 %, pureté 99,9 % | |
| Avec de l'hydroxyde de sodium;Dans l'eau;toluène;à 10 - 55℃ ;pour 11h; | Dans un ballon à trois cols, 97 g de chlorure de p-toluènesulfonyle et 180 ml de toluène ont été placés. Agiter et dissoudre, filtrer, maintenir au chaud à 15-20 C, 60 g de 2-thiophèneéthanol et 60 ml de toluène ont été placés dans un ballon à trois cols.120g d'hydroxyde de sodium aqueux à 40%, sous agitation,Une solution de 97 g de chlorure de p-toluènesulfonyle dissous dans 180 ml de toluène a été ajoutée goutte à goutte à 10-20 C, et le mélange a été agité pendant 3 h, puis chauffé à 45-55 C pendant 8 h, et laissé au repos pour la stratification. La phase organique est dépressurisée pour éliminer le toluène pour donner le p-toluènesulfonate de 2-(2-thiophène)éthanol.Huile brun rougeâtre ;la formule moléculaire est la suivante : | |
| 235,1 g | Avec de l'hydroxyde de sodium;Dans le chloroforme ;à 0 - 5℃ ;pendant 12h; | Au ballon de réaction, l'étape ci-dessus 2-thiophène éthanol : 112 g, chloroforme 600 g, PTSC 249,4 g, et le mélange a été refroidi à 0-5 C.Ajouter 834 g de solution d'hydroxyde de sodium à 10 % en masse, contrôler la température 0 -5 C, maintenir au chaud pendant 12h, ajouter de l'acide chlorhydrique jusqu'à pH=12, couche, la couche d'eau est extraite avec 300g de chloroforme, et la couche de chloroforme est combinée.Après lavage avec 100 g d'eau, la phase organique est distillée à sec pour donner le p-toluènesulfonate de 2-thiophèneéthanol : 235,1 g rendement 95,3 %, pureté 99,5 %. |
Écrivez votre message ici et envoyez-le nous
![[ CAS No. 141109-14-0 ] (S)-Méthyl 2-amino-2-(2-chlorophényl)acétate](https://www.ohlsenwin.com/uploads/Chlorophenylglycine-methyl-ester-2-300x300.png)
![[ N° CAS 141109-19-5 ] Chlorhydrate de 2-(2-chlorophényl)-2-((2-(thiophén-2-yl)éthyl)amino)acétate de (S)-méthyle](https://www.ohlsenwin.com/uploads/Chlorophenylglycine-methyl-ester-31-300x300.png)
![[ N° CAS 40412-06-4 ] 4-méthylbenzènesulfonate de 2-(thiophén-2-yl)éthyle](https://www.ohlsenwin.com/uploads/ethanol-Tosylate-4-300x300.png)